
Servicios

Estudios Observacionales
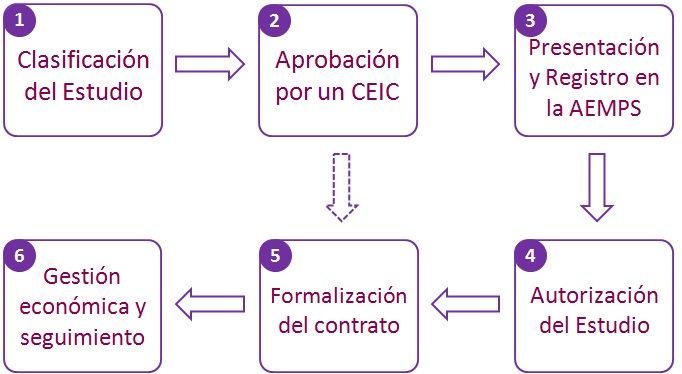
Ruta administrativa de un Estudio Observacional
-
Clasificación del estudio
La Agencia Española del Medicamento y Productos Sanitarios (AMPS) realizará una clasificación previa de todos los estudios clínicos o epidemiológicos no aleatorizados, que se realicen con seres humanos o con registros médicos y que tengan uno o varios medicamentos como exposición de interés. Las categorías son:
-
Estudios Posautorización (EPA) Observacionales.
Estos estudios se consideran necesarios para la obtención de un conocimiento que los Ensayos Clínicos
controlados realizados durante el desarrollo clínico de los medicamentos no aportan. A su vez, pueden agruparse en:
- EPA Ligados a la autorización (EPA-LA). Estudios posautorización cuya realización tiene lugar a instancia de las autoridades reguladoras y ligada a la autorización de comercialización
- EPA Seguimiento prospectivo promovido por Autoridades Sanitarias (EPA-AS) o financiado con fondos públicos.
- Otros EPA de seguimiento prospectivo (EPA-SP) que no corresponden a ninguna de las categorías anteriores.
- EPA otros diseños (EPA-OD). EPA con diseño diferente al de seguimiento prospectivo, por ejemplo, estudios transversales o retrospectivos.
- Estudios Observacionales no EPA. Estudios en los que el factor de exposición fundamental investigado no es un medicamento; por ejemplo estudios de incidencia o de prevalencia de enfermedades, etc.
-
Estudios Posautorización (EPA) Observacionales.
Estos estudios se consideran necesarios para la obtención de un conocimiento que los Ensayos Clínicos
controlados realizados durante el desarrollo clínico de los medicamentos no aportan. A su vez, pueden agruparse en:
-
Aprobación por un CEIC.
Una vez clasificado, es necesario obtener el dictamen favorable del estudio por un Comité de Ética de la Investigación Clínica (CEIC) acreditado en España.
En nuestra Comunidad Autónoma, los comités competentes en esta materia son: el Comite Coordinador de Ética de la Investigación Biomédica de Andalucía (CCEIBA) y los Comités de Ética de la Investigación de Centros que realicen Investigación Biomédica.
Los órganos de ética de la investigación biomédica en Andalucía están regulados en el Decreto 439/2010 de 14 de diciembre.
-
Presentación de la propuesta y registro en la AEMPS.
En función de la clasificación del Estudio que estemos considerando, será necesario:
- EPA-LA. Presentación a la AEMPS y registro en la propia AEMPS.
- EPA-AS. Presentación al Comité de Coordinación del EPA y, tras la evaluación, registro en el AEMPS.
- EPA-SP. Presentación a las Comunidades Autónomas y al AEMPS y registro posterior en el AEMPS.
- EPA-OD. Presentación a la AEMPS, sin ser necesario su registro.
- No-EPA. Los Estudios Observacionales no EPA, no requieren presentación de la propuesta ni registro..
-
Autorización del estudio.
Los Estudios Observacionales del tipo EPA-LA, EPA-AS y EPA-SP necesitan una autorización para poder ser llevados a cabo. Esta autorización debe ser concedida por la AEMPS en el caso de EPA-LA y EPA-AS o por cada Comunidad Autónoma en el caso de los EPA-SP.
-
Formalización del contrato.
Todos los estudios posautorización requerirán para su inicio el oportuno contrato del promotor con los responsables de las entidades proveedoras de servicios sanitarios donde prevea llevarse a cabo el estudio o el visto bueno del director d el centro conforme a los procedimientos específicos que se manejen en cada centro.
-
Gestión económica y seguimiento.
La gestión y el seguimiento económico del proyecto se lleva siempre a cabo por la Fundación Gestora de Referencia.
Para los estudios EPA-LA, EPA-SP y EPA-AS que sean considerados como de seguimiento prospectivo el promotor deberá enviar un informe de seguimiento anual, o antes si así se solicita. Asimismo, el promotor informará de forma inmediata sobre cualquier incidencia relevante (interrupción, problema grave de seguridad, etc.) que pueda producirse en el transcurso del estudio.
Todas estas comunicaciones se presentarán a los órganos competentes de las Comunidades Autónomas involucradas y a la AEMPS.
Puedes ampliar la información remitiéndote a la Orden SAS/3470/2009 de 16 de diciembre, por la que se publican las directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano.

